|
| Appunti universita |
|
|
| Appunti universita |
|
| Visite: 2334 | Gradito: |
Leggi anche appunti:Endocarditi infettiveENDOCARDITI INFETTIVE Le endocarditi infettive sono dovute alla colonizzazione PneumococchiPNEUMOCOCCHI · Pneumococco o streptococco pneumoniae · Cocchi, EctoparassitosiECTOPARASSITOSI Sono parassitosi che si osservano sulla superficie |
 |
 |
FIBROSI CISTICA
La fibrosi cistica è una malattia ereditaria multisistemica del bambino e dell'adulto, caratterizzata principalmente da ostruzione e infezione delle vie aeree e da maldigestione, con le relative conseguenze.
Inizialmente la fibrosi cistica era una patologia di esclusivo interesse pediatrico; con il miglioramento della diagnosi e della terapia l'aspettativa di vita è aumentata di molto, raggiungendo anche l'età adulta avanzata.
La fibrosi cistica è:
Una malattia genetica a carattere autosomico recessivo;
Più frequente nella razza bianca, a prognosi infausta;
Il tratto genetico recessivo a prognosi sfavorevole più comune nei bianchi;
Una malattia cronica, progressiva, evolutiva.
La sua incidenza in Italia è 1:4200 nei bambini nati vivi, ed è per questo la patologia genetica più frequente; sopravvivenza media >30 anni.
È importante, qualora si diagnostichi la FC in un paziente, fare un accurato screening nei parenti più stretti. Se un paziente portatore sano intende intraprendere una gravidanza, sarà necessario effettuare il test di screening anche sul coniuge, così come un'indagine prenatale del feto.
Età media della diagnosi: La diagnosi sulla base di segni e sintomi, quali malassorbimento, tosse, febbriciattola, ritardo della crescita, in genere viene fatta intorno ai 14.5 mesi;
Per i bambini che presentano ileo da meconio la diagnosi viene posta a 0.2 mesi ed è possibile intervenire:
Chirurgicamente;
tramite il Gastrografin® (diatrizoato), un mezzo di contrasto utile sia dal punto di vista diagnostico che terapeutico;
mezzi litici dall'alto e dal basso.
Per i bambini che ricevono lo screening prenatale la diagnosi è posta a 0.5 mesi tramite IRT/DNA: il protocollo prevede il dosaggio IRT (24-72 h) ed in più la ricerca di mutazioni nel DNA. Per IRT si intende il dosaggio del tripsinogeno immunoreattivo nel siero; il cut-off è di 100 μg/ml e valori > 100 μg/ml sono indice di patologia.
Nel 15% dei bambini, sia italiani che americani, essendo casi subclinici, la diagnosi viene posta in ritardo (10 anni).
Gene CFTR: Nel 1989 è stato clonato il gene responsabile della fibrosi cistica sul cromosoma 7, coinvolto nella sintesi di un trasportatore di membrana di 1480 aminoacidi, definito CFTR, che regola l'ingresso e l'uscita del Cloro dalle cellule; esso è presente sulla membrana apicale delle cellule epiteliali delle ghiandole esocrine.
Classificazione delle mutazioni del gene CFTR ed effetti sulla funzione della proteina: Sono oggi note oltre 1500 mutazioni che portano alla diminuita sintesi del trasportatore o alla sua completa assenza o ad un'alterazione della sua funzione.
Pazienti con mutazioni cosiddette "mild", che producono un difetto del canale del Cloro o un difetto di sintesi, hanno valori medi di test del sudore inferiori rispetto a pazienti con mutazioni severe che determinano una assenza della proteina, un difettoso smistamento o un difetto di regolazione.
La delezione di un singolo residuo di fenilalanina a livello dell'aminoacido 508 (ΔF508), che rappresenta la mutazione a maggiore prevalenza, è responsabile dell'elevata incidenza della patologia nel Nord Europa.
La correlazione tra genotipo CFTR e fenotipo clinico, molto complessa, non è prevedibile nel singolo paziente. Le mutazioni etichettate come "gravi" (ΔF508) si associano quasi uniformemente ad insufficienza pancreatica, ma solo in linea generale ad una più rapida progressione della malattia polmonare. Sembra che polimorfismi di un gene modificatore siano responsabili di gran parte della variabilità della progressione della pneumopatia. L'associazione più stretta con forme di malattia più gravi è con una singola variazione nucleotidica nel gene del TGF-β1. Alleli varianti della lectina legante il mannosio, un fattore chiave dell'immunità innata sistemica, si associano a infezioni polmonari più gravi e a una sopravvivenza ridotta.
Diverse mutazioni sono riscontrabili nei pazienti con normale secrezione di cloro nel sudore. Alcuni pazienti con polimorfismi di entrambi i geni CFTR hanno scarse manifestazioni della fibrosi cistica (o addirittura nessuna) fino all'adolescenza o all'età adulta, quando presentano pancreatite, sinusite, bronchiectasie diffuse o infertilità maschile.
Ruolo del CFTR
Favorisce la conduzione di anioni e modifica altri canali;
Controlla la quantità e la composizione delle secrezioni epiteliali come sudore, muco e succo pancreatico;
Altera questi fluidi compromettono la funzione di molti organi: seni paranasali, polmone, pancreas, intestino, ghiandole sudoripare.
Patogenesi: Quattro osservazioni sono di fondamentale importanza dal punto di vista patogenetico:
l'incapacità di eliminare le secrezioni mucose;
uno scarso contenuto d'acqua nelle secrezioni mucose;
un elevato contenuto di sali nel sudore e nelle secrezioni sierose;
infezioni croniche limitate all'apparato respiratorio.
![]()
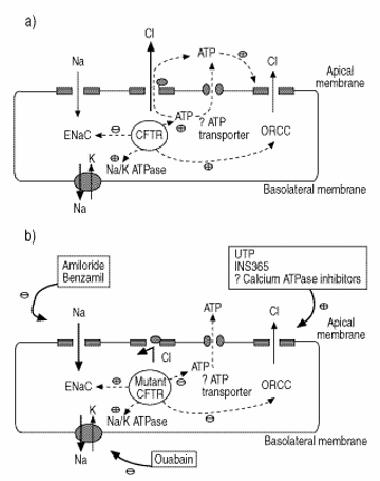 Inoltre, vi è una maggiore differenza di
potenziale negativo attraverso l'epitelio respiratorio nei pazienti con fibrosi
cistica rispetto ai soggetti di controllo. In questi pazienti sono state
dimostrate proprietà elettriche aberranti anche nell'epitelio duttale delle
ghiandole sudoripare; le membrane delle cellule epiteliali non sono in grado di
secernere ioni cloro in risposta a segnali mediati dal cAMP e, almeno nel
tratto respiratorio, viene assorbita una quantità eccessiva di sodio attraverso
queste membrane. Tali difetti sono tutti ascrivibili a una disfunzione del
CFTR.
Inoltre, vi è una maggiore differenza di
potenziale negativo attraverso l'epitelio respiratorio nei pazienti con fibrosi
cistica rispetto ai soggetti di controllo. In questi pazienti sono state
dimostrate proprietà elettriche aberranti anche nell'epitelio duttale delle
ghiandole sudoripare; le membrane delle cellule epiteliali non sono in grado di
secernere ioni cloro in risposta a segnali mediati dal cAMP e, almeno nel
tratto respiratorio, viene assorbita una quantità eccessiva di sodio attraverso
queste membrane. Tali difetti sono tutti ascrivibili a una disfunzione del
CFTR.
Poiché l'acqua segue il movimento dei Sali, il flusso netto d'acqua previsto andrà dal lume delle vie aeree alla sottomucosa e sarà maggiore attraverso l'epitelio con fibrosi cistica. L'aumentato riassorbimento di Na+ da parte delle cellule, nella fibrosi cistica, si associa a un aumento della conduttanza per il Na+ sensibile all'amiloride attraverso la membrana apicale (luminale) e a un aumento dei siti Na+-K+ATPasi sulla membrana basolaterale. La conduttanza del Cl− cAMP-mediata della membrana apicale, associata al regolatore trans membrana della FC (CFTR), non è funzionale nell'epitelio dei pazienti con fibrosi cistica; in compenso, è presente una conduttanza del Cl− alternativa, attivata dal calcio, nelle cellule normali e in quelle con FC. Si postula che nella FC le cellule abbiano una capacità limitata di secernere il Cl− e riassorbano il Na+ in quantità eccessiva, limitando la disponibilità di acqua per idratare secrezioni che quindi possono essere eliminate dal lume delle vie aeree.
Alterazioni della funzione di CFTR
![]()
 Difettoso assorbimento di NaCl con aumento della
concentrazione di cloro nel sudore (Cl−>60
mmol/litro);
Difettoso assorbimento di NaCl con aumento della
concentrazione di cloro nel sudore (Cl−>60
mmol/litro);
Ridotta regolazione del trasporto di volumi isotonici a livello polmonare con bronchiectasia, pneumotorace, emottisi e cuore polmonare;
fegato: malattia ostruttiva delle vie biliari;
Ridotta secrezione di Na+HCO3− a livello del pancreas: diabete mellito insulino-dipendente e insufficienza enzimatica;
Ridotta secrezione di cloruri nel piccolo intestino: ileo da meconio;
organi genitali: infertilità maschile e essenza congenita dei dotti deferenti.
Le secrezioni degli organi coinvolti sono molto dense, andando a determinare apoptosi delle cellule.
![]()
 Patogenesi delle alterazioni polmonari: La fisiopatologia che è stata postulata per
l'epitelio delle vie aeree comprende l'incapacità di secernere sali e,
secondariamente, acqua, in presenza di un eccessivo riassorbimento di entrambi.
La conseguenza è che la quantità di acqua sulla superficie delle vie aeree
risulta insufficiente a idratare le secrezioni; queste ultime divengono quindi
più viscose ed elastiche (gommose) e sono più difficili da eliminare da parte
del sistema muco ciliare e degli altri meccanismi. L'alterata reologia del muco
può essere aggravata da basse concentrazioni di HCO3−
e da un pH più acido. Il ristagno di queste secrezioni causa ostruzione delle
vie aeree a partire da quelle di calibro minore, i bronchioli. L'ostruzione al
flusso a livello delle piccole vie aeree è l'anomalia fisiologica più precoce,
osservabile nell'apparato respiratorio.
Patogenesi delle alterazioni polmonari: La fisiopatologia che è stata postulata per
l'epitelio delle vie aeree comprende l'incapacità di secernere sali e,
secondariamente, acqua, in presenza di un eccessivo riassorbimento di entrambi.
La conseguenza è che la quantità di acqua sulla superficie delle vie aeree
risulta insufficiente a idratare le secrezioni; queste ultime divengono quindi
più viscose ed elastiche (gommose) e sono più difficili da eliminare da parte
del sistema muco ciliare e degli altri meccanismi. L'alterata reologia del muco
può essere aggravata da basse concentrazioni di HCO3−
e da un pH più acido. Il ristagno di queste secrezioni causa ostruzione delle
vie aeree a partire da quelle di calibro minore, i bronchioli. L'ostruzione al
flusso a livello delle piccole vie aeree è l'anomalia fisiologica più precoce,
osservabile nell'apparato respiratorio.
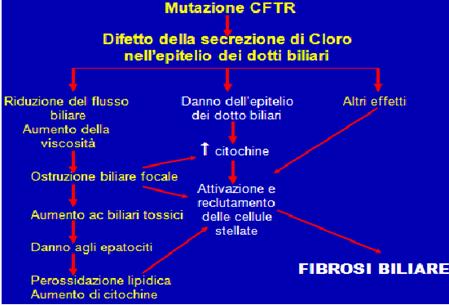
È plausibile che eventi fisiopatologici analoghi abbiano luogo nei dotti pancreatici, nei doti biliari e nei vasi deferenti, con conseguente disidratazione delle secrezioni proteinacee e ostruzione.
L'incapacità di eliminare rapidamente i batteri inalati, la loro conseguente colonizzazione persistente e la risposta infiammatoria della parete delle vie aeree, rappresentano la spiegazioni più verosimili all'infezione cronica che sempre si associa alla fibrosi cistica e che rimane limitata l'apparato respiratorio.
La compromissione polmonare può essere alterata da fattori ambientali e polimorfismi di altri geni relativi ad altri organi (geni modificatori).
Principali manifestazioni cliniche
Malattia polmonare ostruttiva cronica, generalmente associata a infezione cronica da Stafilococcus Aureo o Pseudomonas Aeruginosa (>95%); la prevalenza di infezioni da Pseudomonas Aeruginosa aumenta all'aumentare dell'età, ed è il tipo di infezione più problematica; la prevalenza d'infezione da Stafilococcus Aureus, invece, diminuisce con l'età. Le prime manifestazioni polmonari sono la bronchiolite e la bronchite cronica, ma dopo mesi o anni le alterazioni strutturali dalla parete delle vie aeree provocano la formazione di bronchiolectasie e bronchiectasie. La tosse è il sintomo più costante del coinvolgimento polmonare: dapprima secca e stizzosa, può poi divenire grassa e produttiva. Il muco espettorato è di solito purulento. Alcuni pazienti restano asintomatici per lunghi periodi o sembrano avere infezioni respiratorie acute prolungare ma intermittenti; altri sviluppano una tosse cronica nella prima settimana di vita o sviluppano polmoniti ripetute;
Sinusite cronica (>95%); spesso i pazienti presentano anche polipi indotti dall'infiammazione della mucosa;
Insufficienza pancreatica esocrina (~85%), i cui sintomi comprendono evacuazioni frequenti di feci abbondanti ed oleose e mancato aumento ponderale;
i segni fisici tipici sono addome globoso, riduzione della massa muscolare, crescita stentata e ritardo della maturazione. All'insufficienza pancreatica frequentemente si associa distruzione delle cellule beta con comparsa di diabete;
Azoospermia ostruttiva (>95% dei maschi), con funzione sessuale generalmente intatta; amenorrea secondaria nelle adolescenti;
Malattia epatobiliare (~5% per la cirrosi biliare associata a ipertensione portale; 10-40% con anomalia biochimiche, aspecifiche, come ad esempio aumento delle transaminasi che rispondono bene alla terapia con acido ursodesossicolico), le cui manifestazioni cliniche comprendono ittero, ascite, ematemesi da varici esofagee, e segni di ipersplenismo;
Ileo da meconio (nel neonato 10-20%).
 Diagnosi: Si può fare diagnosi di FC sulla base di riscontri
clinici, quali familiarità per la malattia, Pneumopatia cronica, Insufficienza
pancreatica, Ileo da meconio e la positività al test del sudore.
Diagnosi: Si può fare diagnosi di FC sulla base di riscontri
clinici, quali familiarità per la malattia, Pneumopatia cronica, Insufficienza
pancreatica, Ileo da meconio e la positività al test del sudore.
![]() Il
Test del sudore è un'analisi quantitativa o qualitativa del sudore per la
determinazione della concentrazione degli elettroliti, della conduttività o
dell'osmolarità.
Il
Test del sudore è un'analisi quantitativa o qualitativa del sudore per la
determinazione della concentrazione degli elettroliti, della conduttività o
dell'osmolarità.
Si stimola per circa 5 minuti la produzione di sudore (sono necessari almeno 75 mg di sudore) mediante ionoforesi pilocarpinica: si applicano sulla cute non infiammata dell'avambraccio o della gamba due elettrodi dotati di sistemi di protezione, su cui trovano posto due tamponi assorbenti o due gel contenenti pilocarpina. L'applicazione di corrente elettrica a basso voltaggio (3mA) favorisce il trasporto della pilocarpina negli strati superficiali della pelle, dove stimola la produzione del sudore; viene assorbito con garza e carta da filtro per 30 minuti, si pesa e il campione viene considerato positivo se supera i 75 mg, meglio se arriva a 100 mg.
La CF Foundation USA raccomanda la valutazione della concentrazione di cloro nel sudore, con o senza la contemporanea valutazione della concentrazione di sodio.
Il test è positivo se i valori di cloro sono > 60mEq/l (o <40). Negativo se < 40mEq/l (o <30). Border line se compreso tra 40 e 60 mEq/l (o <40 e >30) e richiede una ripetizione del test.
Criteri per la diagnosi: In presenza di un Fenotipo FC positivo e/o FC in un fratello o in una sorella e/o ipertripsinemia neonatale in presenza di un test del sudore positivo e/o due mutazione del gene CFTR e/o NPD (Anomalia della differenza di potenziale nasale). L'NDP è un test ultra specialistico che si fa solo in alcuni centri.
La misura si effettua utilizzando da un lato un elettrodo esploratore sulla mucosa nasale ed un elettrodo di riferimento posto sulla cute (in genere di un braccio): la differenza di potenziale elettrico tra i due poli è quella che esprime lo stato di funzionamento dei canali che provvedono a livello di mucosa al trasporto di elettroliti. Nella fibrosi cistica il difetto di proteina CFTR fa si che vi sia scarsa secrezione di cloro, e quindi una sua bassa concentrazione sulla superficie mucosa, ed un elevato riassorbimento di sodio attraverso il canale del sodio, con conseguente sua bassa concentrazione sulla mucosa.
Terapia: approccio multidisciplinare.
Fisioterapia respiratoria: va effettuata da un fiosioterapista specializzato in FC; il suo obiettivo è di mobilizzare i muchi che sono adesi alle pareti dei bronchi tramite la manovra del picchiettamento (si basa sul principio secondo cui se la tosse libera le vie aeree principali dal muco, sono necessarie vibrazioni per rimuovere le secrezioni dalle vie aeree di piccolo calibro, dove le velocità di flusso espiratorio sono inferiori); induce quindi il paziente a sputare, in modo da favorirne l'espulsione; in seguito passa a manovre di ginnastica respiratoria con particolari mascherine e apparecchi che favoriscono la capacità respiratoria del paziente;
Esercizio fisico;
Aereosol terapia con uso adeguato di antibiotici per via sistemica o topica;
Nutrizione adeguata: ipercalorica e con vitamine liposolubili;
Supplementazione con estratti pancreatici (da somministrare ad ogni pasto): riducono ma non correggono completamente le perdite di grassi e azoto, il dosaggio e il prodotto devono essere individualizzati per ogni paziente; le microsfere enterorivestite contenenti enzimi, sensibili al pH, sono oggi le più utilizzate; poiché la somministrazione di dosi troppo elevate è stata correlata a stenosi coliche tali da richiedere l'intervento chirurgico, la sostituzione enzimatica in genere non dovrebbe superare le 2500 unità di lipasi/kg/pasto;
Vitamine liposolubili (a causa del malassorbimento vengono date per via parenterale o in forte eccesso rispetto al fabbisogno o in formulazioni con polietilenglicole che le rende assorbibili come se fossero vitamine idrosolubili) e minerali, soprattutto zinco e ferro;
Dosaggio ematico di vitamina A, E, B; per la valutazione della vitamina K si ci regola in base al tempo di protrombina;
Estratti pancreatici;
Antiacidi;
![]()
 Acido ursodesossicolico;
Acido ursodesossicolico;
Supporto psicologico.
Obiettivi della terapia
Prevenzione della malattia polmonare;
Controllo dello stato di nutrizione;
Controllo delle complicanze;
Migliorare la qualità di vita dei pazienti.
Terapia della malattia polmonare
Obiettivi specifici: Fluidificazione delle secrezioni con aereosol terapia con liquidi, mucolitici, broncodilatatori, cortisonici, antibiotici (secondo protocolli accreditati); la DNasi umana ricombinante (2.5 mg) somministrata per aeresol, migliora la funzione polmonare, riduce il numero di esacerbazioni polmonari e promuove un senso di benessere;
Fisioterapia respiratoria secondo metodiche specifiche per età
Trattamento delle infezioni: Antibioticoterapia (secondo modalità definite);
Terapia antiinfiammatoria: Farmaci antiinfiammatori.
Problematiche da affrontare
Tosse;
Necessità di fluidificare le secrezioni;
Malnutrizione e mal digestione;
Integrazione medico specialista e medico curante;
Aderenza al trattamento;
Difficoltà di interventi a domicilio;
Difficoltà di accesso e inserimento nel lavoro;
Difficoltà nella procreazione;
Costanza delle cure prestate;
Supporto psicologico;
Infezioni da parte di germi multi resistenti (a causa delle resistenze spesso i pazienti vengono trattati con farmaci nuovi, ancora in via di sperimentazione; se diventano resistenti o se si sensibilizzano al farmaco vengono sottoposti a desensitizzazione, in modo da poter essere nuovamente sottoposti al farmaco evitando il rischio di gravi effetti collaterali);
Diabete, osteoporosi, cirrosi (la si può prevenire con la somministrazione di acido ursodesossicolico, acido biliare "buono", che ha un effetto coleretico ed impedisce quindi il ristagno di bile).
Discinesia ciliare primitiva
Disordine ereditario a trasmissione autosomica recessiva con prevalenza stimata nella popolazione approssimativamente di 1/20.000 nati vivi. 50% sono affetti da situs inversus.
Patogenesi: le cilia sono estroflessioni della superficie apicale della membrana plasmatica dell'epitelio della mucosa delle vie aeree (superiori ed inferiori) e partecipano ai meccanismi di difesa aspecifici dell'albero tracheobronchiale mediante la rimozione continua del materiale estraneo con la clearance muco-ciliare. Nella DCP si manifestano diverse alterazioni ciliari:
Assenza dei bracci di dineina;
Assenza dei ponti radiali;
Assenza dei ponti di nexina;
Assenza dei microtubuli centrali con trasposizione centrale di una coppia periferica.
Clinica: può manifestarsi alla nascita con distress respiratorio neonatale, ma più spesso si rende evidente nelle prime settimane di vita con rinite mucopurulenta e tosse catarrale persistente. Nelle età successive si manifestano complicanze alle vie aeree superiori (otite media e sinusite cronica), inferiori (bronchite cronica e polmoniti ricorrenti) nonché bronchiectasie.
Diagnosi
Analisi ultrastrutturale delle ciglia in microscopia elettronica;
Test alla saccarina (bambini collaboranti);
Valutazione diretta al microscopio ottico e videoregistrazione del battito ciliare.
Diagnosi differenziale
Asma bronchiale;
Reflusso gastro-esofageo;
Fibrosi cistica;
Deficit immunitari.
Terapia: evitare il ristagno di muco (fisioterapia respiratoria), trattamento antibiotico delle riacutizzazioni infettive, astensione dal fumo, vaccinazione antinfluenzale e antipneumococcica.
Asma bronchiale
L'asma è una malattia infiammatoria cronica caratterizzata da episodi ricorrenti o persistenti di broncospasmo che si manifestano con wheezing (respiro sibilante), dispnea, tachipnea, tosse particolarmente notturna o dopo esercizio fisico e senso di costrizione toracica; ostruzione bronchiale generalmente reversibile spontaneamente o dopo trattamento farmacologico; iperreattività bronchiale; infiltrazione di cellule infiammatorie, rilascio di mediatori e rimodellamento strutturale delle vie aeree. La gravità delle manifestazioni cliniche dell'asma é solitamente correlata all'entità dell'ostruzione bronchiale, ma può essere percepita in maniera differente da diversi individui o nelle diverse fasi della malattia.
Epidemiologia: negli ultimi 20 anni la prevalenza di asma è aumentata considerevolmente soprattutto nei bambini in molti Paesi, anche se negli ultimi anni il trend dell'asma negli adulti non è in ulteriore aumento in parecchie nazioni (Inghilterra, Italia, Svizzera, Australia, Messico). Attualmente il trend è in diminuzione nei bambini (Inghilterra, Australia). Nella prima infanzia la prevalenza è maggiore tra i maschi per il minor calibro delle vie aeree, poi la tendenza si inverte.
![]()
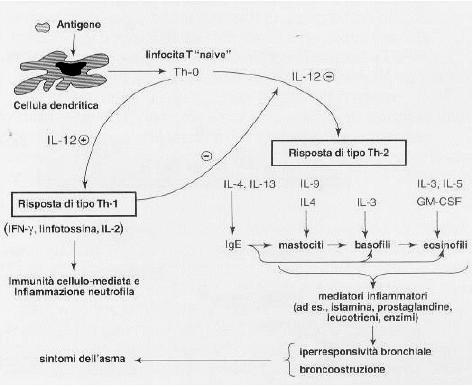 Patogenesi: l'insorgenza dell'asma è dovuta ad un insieme di fattori
genetici legati all'iperresponsività bronchiale o ad una storia di atopia
(predisposizione genetica a produrre IgE eccessive) ma anche a fattori quali lo
status sociale, l'uso di farmaci, il fumo in famiglia, la vita in città e comunque
l'esposizione ad allergeni inalatori o sostanze irritanti.
Patogenesi: l'insorgenza dell'asma è dovuta ad un insieme di fattori
genetici legati all'iperresponsività bronchiale o ad una storia di atopia
(predisposizione genetica a produrre IgE eccessive) ma anche a fattori quali lo
status sociale, l'uso di farmaci, il fumo in famiglia, la vita in città e comunque
l'esposizione ad allergeni inalatori o sostanze irritanti.
Molti mediatori dell'infiammazione nelle secrezioni delle vie aeree dei pazienti asmatici contribuiscono alla bronco-costrizione, alla secrezione di muco e allo stravaso nel microcircolo.
Lo stravaso, una componente costante delle reazioni infiammatorie, porta a un edema della sottomucosa, aumenta la resistenza delle vie aeree e contribuisce all'iperreattività.
I mediatori comprendono l'istamina e i prodotti del metabolismo dell'acido arachidonico (i leucotrieni e i trombossani, entrambi in grado di aumentare transitoriamente l'iperreattività delle vie aeree). I cisteinil-leucotrieni, il LTC4 e il LTD4, sono i più potenti broncocostrittori mai studiati nell'uomo. L'attivazione della risposta allergica da parte delle cellule T è un evento chiave. Le cellule T e i loro prodotti di secrezione (le citochine) mantengono l'infiammazione delle vie aeree. Le citochine prodotte da un gruppo specifico di linfociti, le cellule T CD4+ Th2, favoriscono la crescita e la differenziazione delle cellule, le attivano e le inducono a migrare nelle vie aeree, dove ne prolungano la sopravvivenza. Le principali citochine coinvolte comprendono l'interleuchina (IL)-4, che è necessaria per la produzione di IgE; l'IL-5, che è un fattore chemiotattico per gli eosinofili e il fattore stimolante le colonie di granulociti-macrofagi, simile all'IL-5 per i suoi effetti sugli eosinofili ma meno potente.
Diagnosi: la diagnosi di asma è soprattutto clinica, suggerita da una storia di episodi ricorrenti di respiro sibilante, tosse e dispnea e avvalorata da una storia personale o familiare di malattia atopica (eczema, rinite o congiuntivite allergica, asma). Nel bambino in età prescolare il sintomo principale può essere rappresentato dalla sola tosse notturna.
L'EO può essere negativo o mostrare sibili e fischi telespiratori o sia espiratori che inspiratori; può poi esservi il silenzio respiratorio.
Le prove di funzionalità respiratoria confermano il sospetto diagnostico: in caso di ostruzione alla spirometria si effettuerà il test di reversibilità; in caso di negatività con forte sospetto si effettuerà il test di provocazione bronchiale aspecifico.
In caso di conferma diagnostica si passerà a valutare i fattori di rischio tramite prove allergologiche, indagini per rilevare eventuale reflusso gastroesofageo e/o riniti per ridurre l'esposizione.
Diagnosi differenziale
sindrome da inalazione;
corpo estraneo;
displasia broncopolmonare;
reflusso gastroesofageo;
anelli vascolari - arco aortico destroposto;
tracheobroncomalacia;
disfunzione delle corde vocali;
immunodeficienza primitiva;
fibrosi cistica - discinesia ciliare primitiva;
edema polmonare.
 Terapia: innanzitutto è necessario ridurre la frequenza e la
gravità degli attacchi riducendo dell'esposizione agli allergeni, al fumo, allo
smog e prevenendo le infezioni delle vie respiratorie.
Terapia: innanzitutto è necessario ridurre la frequenza e la
gravità degli attacchi riducendo dell'esposizione agli allergeni, al fumo, allo
smog e prevenendo le infezioni delle vie respiratorie.![]() I farmaci
per l'attacco acuto sono:
I farmaci
per l'attacco acuto sono:
ß2-agonisti inalatori a rapida azione: broncodilatatori con durata di azione di 4-6 ore, rilasciano la muscolatura liscia della parete bronchiale, aumentano la clearance mucociliare, riducono la permeabilità vascolare e possono inibire il rilascio di mediatori dai mastociti. Rappresentano il trattamento di scelta nelle riacutizzazioni d'asma e sono utilizzati per prevenire l'asma associata all'esercizio fisico;
Glucocorticosteroidi sistemici;
Anticolinergici inalatori: sono broncodilatatori che bloccano gli effetti dell'acetilcolina rilasciata dalle vie efferenti postgangliari vagali e inducono broncodilatazione riducendo il tono intrinseco vagale nelle vie aeree, ma non hanno alcun effetto sull'infiammazione. Rispetto ai farmaci β2-agonisti per via inalatoria, gli anticolinergici hanno un minor effetto broncodilatatore ed agiscono più lentamente (effetto massimo dopo 30-60 minuti). Nel trattamento delle riacutizzazioni asmatiche avrebbe un'azione additiva quando nebulizzato insieme ai β2-agonisti a rapida insorgenza d'azione; si usa come farmaco broncodilatatore alternativo per i pazienti che lamentano effetti collaterali da assunzione di β2-agonisti a rapida insorgenza d'azione, come tachicardia, aritmia e tremori;
Metilxantine ad azione rapida (teofillina a breve durata d'azione): farmaco antiasmatico sintomatico usato come broncodilatatore aggiuntivo nell' asma grave. L'insorgenza d'azione è più ritardata rispetto ai β2-agonisti a rapida insorgenza d'azione, sono utilizzati per mantenere la risposta ai SABA nell'intervallo tra le somministrazioni.
La terapia di mantenimento consiste in:
Glucocorticosteroidi inalatori: sono i più potenti agenti antinfiammatori, attivi topicamente, poco assorbiti, e dunque meno in grado di causare effetti avversi. Riducono il bisogno degli steroidi sistemici e sono utilizzati per trattamenti a lungo termine per il controllo dei sintomi e soppressione dell'infiammazione (inibiscono la produzione di citochine, l'attivazione delle molecole di adesione, la migrazione ed attivazione delle cellule della flogosi). Bloccano la fase tardiva della risposta asmatica all'allergene e riducono l'iperreattività bronchiale;
ß-agonisti inalatori a lunga durata d'azione: broncodilatatori con durata d'azione superiore alle 12 ore. Prevenzione dell'asma notturna o associata all'esercizio fisico, utilizzati in associazione a glucocorticoidi per via inalatoria quando le dosi standard di steroidi per via inalatoria falliscono nel raggiungere il controllo dell'asma;
Antagonisti recettoriali dei leucotrieni (montelukast): modesto effetto antinfiammatorio, si usa in terapia aggiuntiva agli steroidi per via inalatoria e nella prevenzione dell'asma da sforzo;
Glucocorticosteroidi orali: sono utilizzati sia nel controllo dell'asma grave persistente (terapia di fondo) che nel trattamento delle riacutizzazioni (terapia sintomatica). Sebbene come sintomatici l'azione non sia immediata (dopo 3-4 ore dalla somministrazione), sono egualmente importanti in quanto ne facilitano la risoluzione, prevengono le recidive, riducono il ricorso al ricovero e la mortalità;
Metilxantine a lento rilascio (orali o parenterali): broncodilatatore che può avere effetti antinfiammatori. L'effetto broncodilatatore è legato alla inibizione della fosfodiesterasi ed è rilevabile ad alte concentrazioni mentre l'effetto antinfiammatorio è dovuto ad un meccanismo sconosciuto e si verifica a concentrazioni più basse. Sono utilizzati per la prevenzione e il controllo a lungo termine dei sintomi broncospastici, in particolare dell'asma notturno;
Cromoni inalatori: bloccano la fase precoce e tardiva dell'asma; interferiscono con i canali del cloro, inibiscono il rilascio da parte dei mastociti di mediatori IgE-mediati, inibiscono l'attivazione e il rilascio dei mediatori dagli eosinofili e cellule epiteliali e la risposta acuta all'aria fredda ed all'esercizio fisico.
Nel trattamento dell'asma nell'infanzia si deve tenere conto che il piccolo che abbia respiro sibilante non sempre è asmatico:
transient early wheezers: prevalenti fino ai 3 anni, sono piccoli che spesso hanno avuto difficoltà respiratorie alla nascita, figli di madri giovani e fumatrici, prematuri e senza storia di atopia;
non atopic wheezers: 3-6 anni, associati a infezione da VRS, il quadro simil-asmatico si risolve intorno ai 13 anni. Non hanno storia di atopia;
atopic wheezers: sono i veri asmatici, in cui la patologia può esordire prima di 3 o dopo 3 anni; prima dei 3 anni sono comunque una minoranza tra i bambini che presentano respiro sibilante
Attualmente, i glucocorticoidi per via inalatoria sono i farmaci di fondo più efficaci e sono raccomandati per l'asma persistente ad ogni livello di gravità: il trattamento a lungo termine con glucocorticoidi per via inalatoria riduce considerevolmente la frequenza e la gravità delle riacutizzazioni e non ha dimostrato alcuna associazione con l'aumento dell'osteoporosi o delle fratture ossee; studi condotti su più di 3500 bambini, trattati con glucocorticoidi per via inalatoria a dosaggi medio-bassi per periodi di 1-13 anni, non hanno dimostrato alcun effetto negativo sulla statura definitiva.
Per la somministrazione si raccomanda:
<4 anni: aerosol in bombolette pressurizzate con camera di espansione e maschera facciale;
4-6 anni: aerosol in bombolette pressurizzate con camera di espansione con boccaglio o maschera facciale;
>6 anni: aerosol in bombolette pressurizzate con camera di espansione con boccaglio o erogatore di polvere.
Il salmeterolo è autorizzato dopo i 4 anni, il formoterolo dopo i 6.
Attacco acuto di asma
lieve: assente o lieve impegno dei muscoli accessori, normale capacità a parlare, saturazione di ossigeno >95%;
medio: modesto uso dei muscoli accessori con rientramenti toracici, limitazione a parlare, saturazione di ossigeno tra 92-95%;
severo: agitazione, notevole impegno dei muscoli accessori con marcati rientramenti, inabilità a parlare, frequenza respiratoria >30 atti/min (>50 atti/min nei bambini di età <5 anni), frequenza cardiaca >120 battiti/min (>160 battiti/min nei lattanti), saturazione di ossigeno <92%, all'auscultazione i sibili possono essere addirittura assenti.
Trattamento: ossigeno per raggiungere una SatO2 > 92%, ß2 agonisti (salbutamolo), anticolinergici, steroidi, aminofillina.
|
| Appunti su: meql unitC3A0 di misura test sudore valori riferimento, |
|
| Appunti Bambini |  |
| Tesine Bellezza |  |
| Lezioni Nutrizione |  |