|
| Appunti scientifiche |
|
|
| Appunti scientifiche |
|
| Visite: 5850 | Gradito: |
Leggi anche appunti:I primi modelli subatomici: Dalton, Thomson e RutherfordI primi modelli subatomici: Dalton, Thomson e Rutherford Nel corso degli Nomenclatura complessiNomenclatura complessi I metalli di transizione, che allo stato elementare EteriEteri Come si è già visto [147] sono composti di formula generale R - O |
 |
 |
Reattività del benzene






L'attacco dell'elettrofilo su un benzene monosostituito avviene su una delle 5 posizioni libere, che NON sono equivalenti. Rispetto al sostituente X già presente sull'anello si distinguono 2 posizioni orto, 2 posizioni meta e 1 posizione para. In relazione all'effetto elettronico del sostituente X possono essere privilegiate le posizioni orto,para, oppure la posizione meta. Inoltre, il sostituente X può rendere le posizioni orto/para o meta più o meno reattive rispetto a una posizione del benzene non sostituito. Si hanno sostituenti X attivanti orto/para orientanti, oppure disattivanti orto/para orientanti, oppure disattivanti meta orientanti.


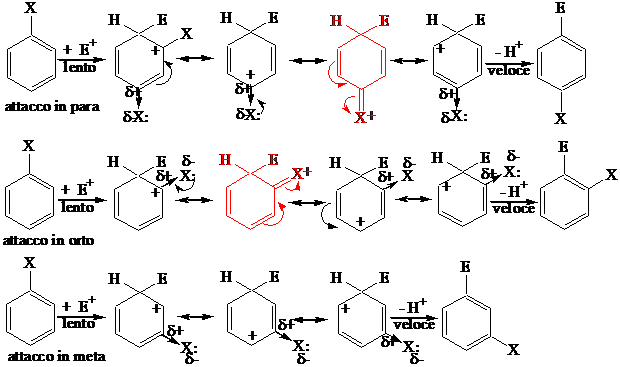

2) La sostituzione nucleofila aromatica

A differenza dei legami multipli CC di alcheni e alchini, il nucleo benzenico (e in generale i nuclei aromatici) subiscono con difficoltà le reazioni di ossidazione e riduzione, che avvengono solo in condizioni drastiche. Le reazioni più interessanti sono:
A) ossidazione di alchil benzeni con KMnO4, che conduce ad acido benzoico indipendentemente dalla lunghezza della catena alchilica

B) Ossidazione dell'idrochinone (p-idrossifenolo) a benzochinone. La reazione è facilmente reversibile e può essere controllata dal pH.

C) Riduzione con sodio metallico a cicloesadiene (Reazione di Birch)
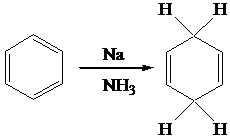
D) Riduzione catalitica a cicloesano. Avviene solo in condizioni molto drastiche.

Sostituzione Elettrofila in Aromatici Eteronucleari
Pirrolo, tiofene e furano subiscono l'attacco nella posizione 2. Sono tutti più reattivi del benzene.

Sostituzione Elettrofila in Aromatici Polinucleari e Polieteronucleari.
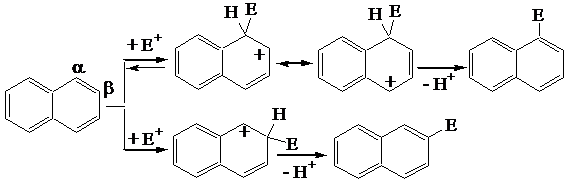
L'isomeri a si forma più velocemente, ma in modo reversibile. Se la reazione è condotta a temperatura bassa (controllo cinetico della reazione) l'isomero a predomina, mente a temperatura alta predomina l'isomero b, termodinamicamente più stabile (controllo termodinamico della reazione).

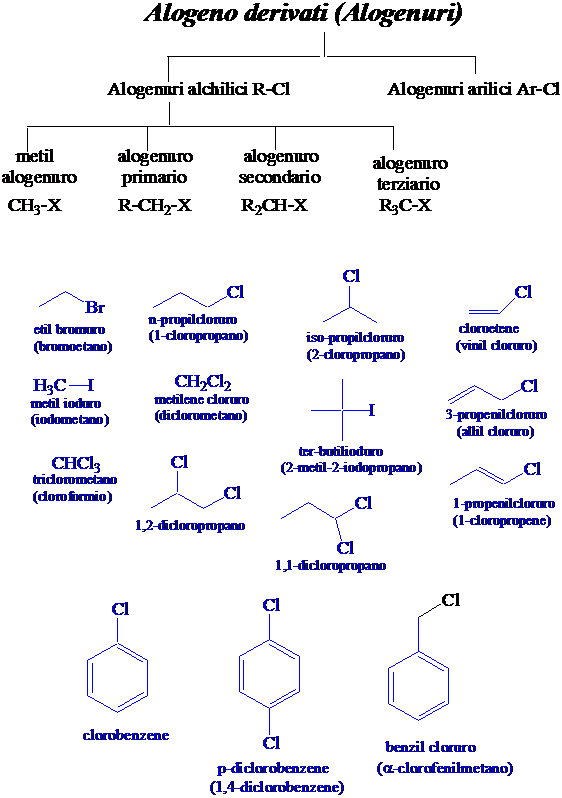
Alchil alogenuri
Da alogenazione radicalica dei corrispondenti alcani (R-H + X2 R-X + HX ; X = Cl, Br; vedi capitolo Reazioni degli alcani)
Da addizione elettrofila di acidi alogenidrici a doppi legami di alcheni (alchene + HX, X = Cl, Br, I; vedi capitolo Reazioni degli alcheni) [Ricorda: considera la regiochimica della reazione, regola di Markovnikov]
Dagli alcoli per trattamento con acidi alogenidrici (ROH + HX; reazione di sostituzione nucleofila alifatica SN1; trattata nel capitolo Reazioni degli alcoli) [Ricorda: la reazione procede via carbocatione; possibilità di trasposizioni]
Dagli alcoli per trattamento con cloruro di tionile o tricloruro di fosforo (ROH + SOCl2 o PCl3; reazione di sostituzione nucleofila alifatica SN2; trattata nel capitolo Reazioni degli alcoli)
Dagli alcoli per trasferimento in tosilati e successivo trattamento con acidi alogenidrici o alogenuri (ROH + Ts-Cl R-OTs; R-OTs + X- R-X; reazione di sostituzione nucleofila alifatica SN2; trattata nel capitolo Reazioni degli alcoli)
Aril alogenuri
Da alogenazione elettrofila aromatica del benzene (Ar-H + X2 + AlX3; vedi capitolo Sostituzioni elettrofile aromatiche)
Da sostituzione nucleofila aromatica via sali di diazonio (Ar-N2+ + X-; vedi capitolo Ammine aromatiche)
1) Reazioni con Nucleofili: Reazione di sostituzione nucleofila alifatica monomolecolare (SN1: reazione non stereospecifica, via carbocatione) e bimolecolare (SN2: reazione stereospecifica).
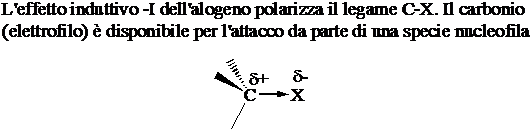
La reazione di sostituzione nucleofila alifatica bimolecolare SN2
Il nucleofilo che entra, Y, (può essere un nucleofilo neutro o uno ione negativo) spinge il nucleofilo che esce, X (effetto push/pull). La reazione avviene in un singolo stadio attraverso uno stato di transizione in cui i tre sostituenti del carbonio si trovano su un piano. La reazione è stereospecifica e avviene con inversione di configurazione sul carbonio che subisce l'attacco.
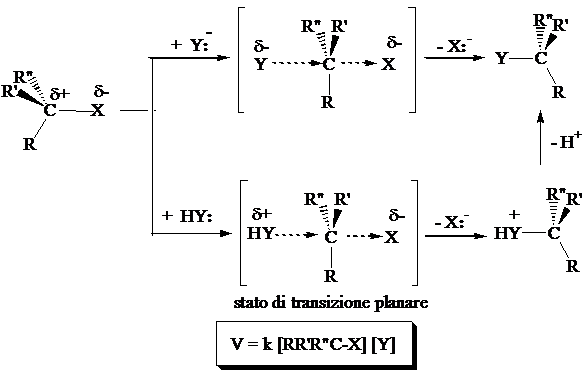
Il solvente strappa il nucleofilo il nucleofilo che esce, X (stadio lento) (effetto pull). Si forma un carbocatione planare che subisce attacco su una o l'altra faccia da parte del nucleofili Y-. La reazione è non-stereospecifica e avviene con racemizzazione .
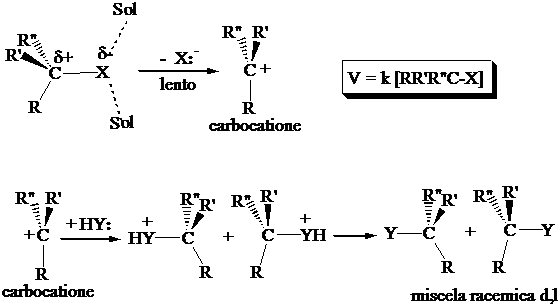
Competizione tra SN1 e SN2
|
Variabile sperimentale |
SN1 |
SN2 |
Esempi |
|
Polarità solvente |
Grande |
Piccola |
CH3CN > CH3COCH3 > CHCl3 > esano |
|
Stabilità carbocatione |
Grande |
Piccola |
PhCH2-X >CH2=CH-CH2-X > R3C-X > R2CH-X > RCH2-X > CH3-X |
|
Dimensioni nucleofilo X o Y |
Grande |
Piccola |
t-BuOH > Bu-OH > MeOH > H2O |
|
Bontà nucleofilo uscente X |
Grande |
Piccola |
I- > Br- > Cl- > F- |
|
Bontà nucleofilo entrante Y |
Piccola |
Grande |
I- > Br- > Cl- > F- /R2S: > R3N: > R2O: / RO- > R2O |

2) Reazioni con basi Reazione di eliminazione monomolecolare (E1: reazione regiospecifica non-stereospecifica) e bimolecolare (E2: reazione trans-stereospecifica e regiospecifica)
Se l'alogenuro possiede idrogeni in b, la specie Y- può comportarsi da base e strappare il protone. Il doppietto di elettroni del legame C-H si ribalta e spinge via il nucleofilo X. La reazione avviene in un singolo stadio. La reazione è trans-stereospecifica e avviene solo quando l'idrogeno e il nucleofilo si trovano sullo stesso piano, ma in posizioni opposte rispetto al legame CC (anti periplanari). E' favorita rispetto alla SN2 quando si usano basi forti (OH-) o, meglio, forti e stericamente impedite (t-BuO-).

Avviene quando sussistono le condizioni per la formazione del carbocatione. Se il carbocatione possiede idrogeni in b, la specie Y- può comportarsi da base e strappare il protone anzichè legarsi al carbonio. Come tutte le reazioni che passano via carbocatione è non-stereospecifica. E' favorita rispetto alla SN2 quando si usano basi forti (OH-).

Entrambi le reazioni E1 e E2 sono regioselettive. La regiochimica è governata dalla regola di Saitzev: viene eliminato selettivamente l'idrogeno in b che conduce all'alchene più ramificato, più stabile.



Reazione di sostituzione nucleofila aromatica.
Come già visto nel capitolo degli Areni, avviene quando in orto/para sono presenti gruppi EWG che stabilizzano l'anione intermedio.


|
n° atomi C |
R- H |
R- F |
R- Cl |
R- Br |
R- I |
R-OH |
ROR |
RCOR |
R-CHO |
R-COOH |
R-NH2 |
R2NH |
R3N |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
In una serie omologa il punto di ebollizione aumenta al crescere del peso molecolare. In una serie eterologa aumenta all'aumentare della polarità del legame R-X (aumentano le interazioni intermolecolari dipolo-dipolo) e all'aumentare della forza dei (possibili) legami a idrogeno.

Metanolo da CO + 3H2 (gas di acqua) CH3OH
Etanolo da idratazione dell'etilene CH2=CH2 + H2O CH3CH2OH (vedi capitolo Alcheni)
Da idratazione degli alcheni (vedi capitolo Alcheni)
Da idroborazione/ossidazione degli alcheni (vedi capitolo Alcheni)
Da sostituzione nucleofila alifatica di alogenuri (idrolisi) R-Cl + H2O ROH + HCl (vedi capitolo Alogenuri)
Da riduzione di aldeidi o chetoni con NaBH4 (vedi capitolo Aldeidi e chetoni)
Da riduzione di acidi o esteri con LiAlH4 (vedi capitoli Acidi carbossilici e Esteri)
Da aldeidi o chetoni con reagenti di Grignard (vedi capitolo Aldeidi e chetoni)
Sostituzione nucleofila aromatica su clorobenzeni PhCl + KOH (vedi capitolo Sostituzione nucleofila aromatica)
Sostituzione nucleofila aromatica su acidi solfonici PhSO3H + KOH (vedi capitolo Sostituzione nucleofila aromatica)
Sostituzione nucleofila aromatica via sali di diazonio (vedi capitolo Ammine)
Da ossidazione di alcheni con KMnO4 o OsO4 (vedi capitolo Alcheni)
Da idrolisi degli epossidi
![]()
La reattività degli alcoli è caratterizzata dalla presenza del legame O-H e del legame R-O fortemente polarizzati e dalla di due doppietti di elettroni di non legame sull'atomo di ossigeno. Pertanto gli alcoli sono acidi, basi e nucleofili. Inoltre, il carbonio legato all'ossigeno è elettrofilo, ossia disponibile all'attacco da parte di specie nucleofile.

![]()

Alcoli alifatici |
pKa |
Alcoli aromatici |
pKa |
|
H-OH |
|
Fenolo |
|
|
CH3-OH |
|
m-nitrofenolo |
|
|
C2H5-OH |
|
p-nitrofenolo |
|
|
2-propanolo |
|
2,4-dinitrofenolo |
|
|
ter-butanolo |
|
2,4,6-trinitrofenolo (ac. Picrico) |
|
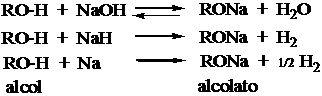

I fenoli e fenati sono nucleofili (e basi) più deboli di alcoli e aalcolati perché il doppietto è delocalizzato sul nucleo benzenico.
Gli alcoli sono substrati per un attacco da parte di nucleofili
Gli alcoli come tali non subiscono mai reazione di sostituzione nucleofila, perché il gruppo -OH non è un gruppo uscente.
![]()
Il gruppo -OH può essere trasformato in buon gruppo uscente nelle seguenti condizioni:
La reazione SN1/E1 in ambiente acido. Gruppo uscente: H2O
Alcoli secondari, terziari, allilici e benzilici formano carbocationi in ambiente acido per protonazione dell'atomo di ossigeno ed eliminazione di acqua. La reazione con acidi alogenidrici conduce alla formazione di alogeno derivati. Con acido solforico si ottiene il prodotto di eliminazione (Reazione di disidratazione degli alcoli)
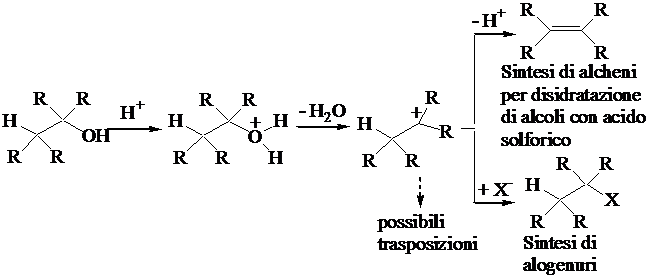
La reazione SN2 con tosilati.
La funzione -OH può essere trasformata in estere dell'acido solfonico (solfonato), che è un bun gruppo uscente in reazioni di SN2. E' molto utilizzata la trasformazione in tosilato, ovvero nell'estere dell'acido para-metilbenzensolfonico, che si ottiene trattando l'alcol con para-metilbenzenesolfonil cloruro (tosil cloruro).
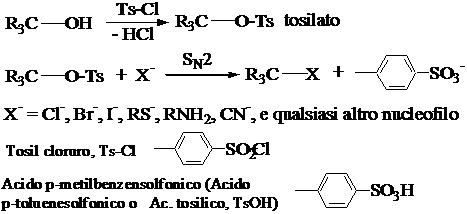
La reazione SN2 con clorosolfiti o clorofosfiti
Reazione molto usata per la sintesi di cloroderivati partendo da alcoli. I clorosolfiti o clorofosfiti si formano come intermedi non isolabili per trattamento dell'alcol con tionil clouro o tricloruro di fosforo. Rispetto alla reazione con HCl ha il vantaggio di non avere reazioni competitive, come la trasposizione o la eliminazione.
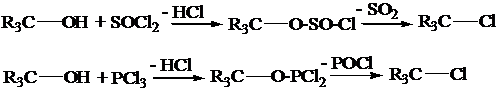
3) I fenoli subiscono sostituzione elettrofila aromatica in orto/para.
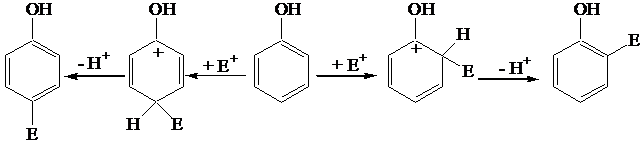

I fenoli sono antiossidanti. Bloccano i radicali attraverso la reazione di idrogeno donazione (reazione di ossidazione), prevenendo l'ossidazione di molecole biologiche come DNA, lipidi, etc. (Il capitolo sugli antiossidanti è trattato in Appendice)

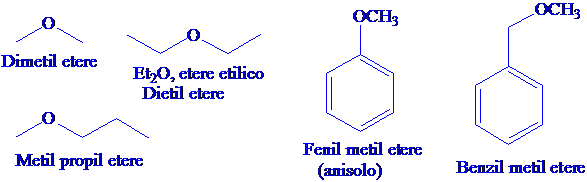
Gli eteri sono composti generalmente poco reattivi, spesso usati come solventi.
Si preparano attraverso la sintesi di Williamson.
![]()
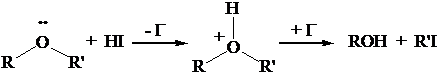
La nomenclatura degli eteri ciclici è definite dale regole IUPAC, anche se per i composti più diffusi si utilizzano i nomi comuni. Tra i principali eteri ciclici meritano una notazione il furano, composto aromatico, il tetraidrofurano (THF), ottimo solvente, e l'ossido di metilene.
|
n |
Saturo |
Insaturo |
|
|
Ossirano (Ossido di metilene) |
Ossirene |
|
|
Ossetano |
Ossete |
|
|
Ossolano (Tetraidrofurano) |
Ossolo (Furano) |
|
|
Ossano |
Ossina (Pirano) |
|
|
Ossepano |
Ossepina |
L'ossido di metilene è il capostipite di una classe di coposti eterociclici ossigenati a 3 termini chiamati epossidi.
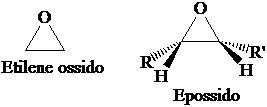
L'ossido di etilene è prodotto industrialmente su larga scala per ossidazione catalica dell'etilene con aria. Il catalizzatore è a base di argento metallico. E' utilizzato come intermedio nella produzione di polimeri poliesteri (PET, Dacron, Mylar, etc).
![]()
Gli epossidi si sintetizzano per ossidazione dei corrispondenti alcheni con peracidi attraverso una reazione stereoselettiva.
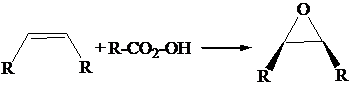
A differenza degli eteri lineari, o degli eteri ciclici a 5 o 6 termini, l'apertura degli epossidi avviene facilmente sia in ambiente acido che basico a causa della notevole tensione d'anello.

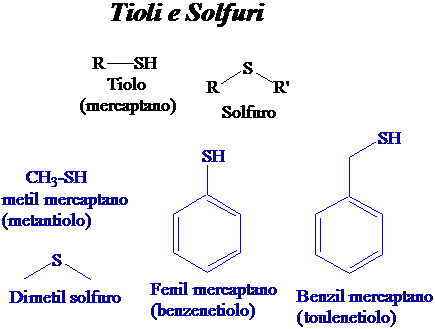
Tioli e solfuri sono caratterizzati da un odore fortemente sgradevole.
I tioli si preparano per reazione SN2 di NaHS sul corrispondente alogeno derivato. Hanno una reattività simile agli alcoli, rispetto ai quali si differenziano per la minor forza del legame S-H e la maggior polarizzabilità del doppietto di elettroni di non legame. Per queste ragioni sono:
acidi più forti. La reazione con una base (es., NaOH) conduce alla formazione dei tiolati.
Basi più deboli.
Nucleofili più forti.
I solfuri si preparano per reazione SN2 tra un tiolato RS- e un alogeno derivato.
Tra i solfuri ciclici merita una notazione il tiofene, composto eteroaromatico di crescente interesse, poiché i suoi polimeri (politiofeni) mostrano spiccate caratteristiche di semiconduttori.
|
n |
Saturo |
Insaturo |
|
|
Tiirano |
Tiirene |
|
|
Tietano |
Tiete |
|
|
Tiolano (Tetraidrotiofene) |
Tiolo (Tiofene) |
|
|
Tiano |
Tiina |
|
|
Tiepano |
Tiepina |
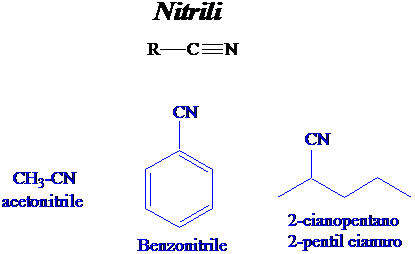
I nitrili alifatici si preparano per reazione SN2 del cianuro di sodio sul corrispondente alogenuro.
![]()
I nitrili aromatici si preparano per reazione di sostituzione nucleofila aromatica via sali di diazonio.
![]()
La reattività è governata dalla forte polarizzazione del legame CN, che subisce facilmente reazione di addizione nucleofila all'atomo di carbonio.
Formazione di ammine per riduzione con LiAlH4

Formazione di acidi carbossilici per idrolisi acida o basica

Formazione di chetoni per reazione con reagenti di Grignard.
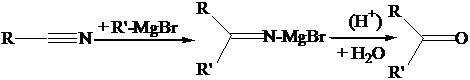
 Proprietà delle ammine
Proprietà delle ammine
La reattività delle ammine è caratterizzata dalla presenza del gruppo NR2, con R = H, alchile o arile.
Il legame N-H è fortemente polarizzato, ma meno del legame O-H. Pertanto le ammine sono acidi più deboli degli alcoli [pka (NH3) = 38]. Viceversa, lo ione ammido è una base più forte dello ione alcolato.
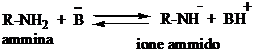
Sull'atomo di azoto è presente un doppietto di non legame. Poiché l'atomo di azoto è meno elettronegativo dell'atomo di ossigeno, le ammine sono nucleofili (reazioni SN2) e basi (formano ioni ammonio) più forti degli alcoli.
3) Il doppietto delle ammine aromatiche è delocalizzato sul nucleo aromatico, quindi meno disponibile. Pertanto le ammine aromatiche sono meno basiche e meno nucleofile delle ammine alifatiche.

Reazione SN2 della ammoniaca su alogenuri alchilici. La reazione è difficilmente controllabile, specialmente quando si vuole sintetizzare una ammina primaria.
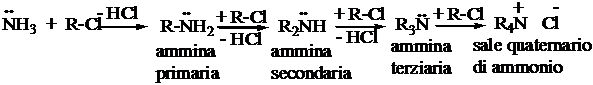
Reazione di sostituzione acilica di ammoniaca su alogenuri acilici e successiva riduzione dell'ammide con idruro di litio alluminio (Vedi anche capitolo "Acidi carbossilici e derivati")
![]()
Reazione SN2 di ioni cianuro su alogenuri alchilici e successiva riduzione del nitrile (Vedi capitolo Nitrili)
![]()

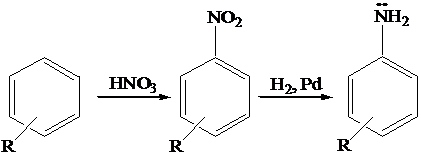
Reazioni delle ammine
1. Formazione di sali in ambiente acido

2. Reazione di SN2 su alogenuri alchilici (Reazione di formazione di ammine)
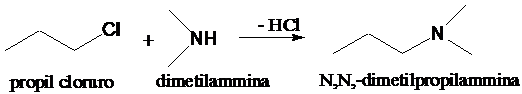
3. Reazione di sostituzione acilica su alogenuri acilici, anidridi o acidi carbossilici (Reazione di formazione di ammidi).
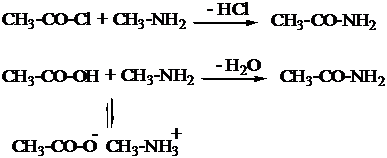
La reazione con acidi carbossilici porta velocemente alla formazione del sale. Per ottenere l'ammide occorre utilizzare un agente disidratante (DCC, dicloesil-carbodiimmide).
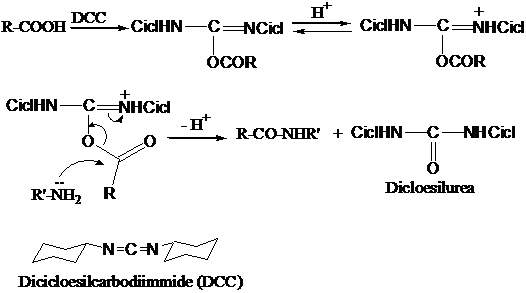
La reazione avviene facilmente anche senza l'utilizzo di DCC quando il gruppo COOH e il gruppo NH2 sono sulla stessa molecola (Reazione intramolecolare di g- o d-amminoacidi). Si ottiene una ammide ciclica (lattame).
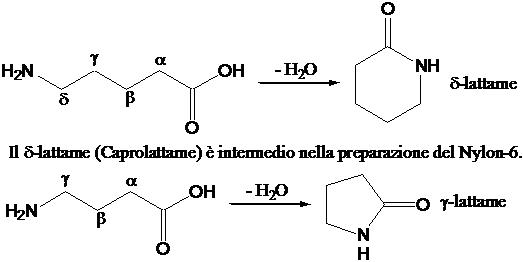
4. La reazione di diazotazione delle ammine aromatiche (Formazione dei sali di diazonio)

I sali di diazonio sono specie instabili e possono decomporre violentemente.
5. La reazione di eliminazione dei sali di ammonio quaternari (Eliminazione di Hoffmann)

Reazione di sostituzione nucleofila aromatica SN1. I sali di diazonio reagiscono con specie nucleofile perdendo una molecola di azoto. La reazione è ampiamente utilizzata per la sintesi di benzeni sostituiti. Lo studio del meccanismo di queste reazioni è abbastanza complesso ed esula dallo scopo di questo corso.

Reazione di copulazione (formazione di azo derivati). I sali di diazonio sono deboli elettrofili e danno reazione di sostituzione elettrofila aromatica su benzeni attivati, come fenoli o aniline. La reazione con fenoli avviene meglio in ambiente basico. Gli azo derivati sono composti fortemente colorati, con tonalità dal giallo al rosso cupo. I coloranti azoici sono azo derivati contenenti opportuni sostituenti capaci di legarsi alle fibre dei tessuti.
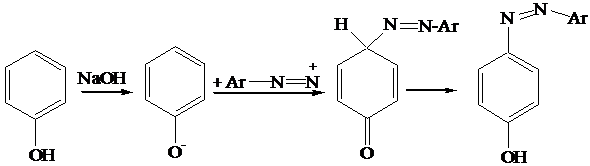

La reattività dei composti carbonilici è governata dalla presenza
del doppio legame C=O (Carbonile) fortemente polarizzato.
L'atomo di carbonio è fortemente elettrofilo e subisce facilmente
attacco da parte di specie nucleofile.

Reattività dei composi carbonilici
Le aldeidi e i chetoni subiscono reazione di addizione nucleofila al carbonile. Se il nucleofilo non è molto forte, la reazione può essere accelerata da catalisi acida: il protone si lega all'ossigeno aumentando la carica positiva sull'atomo di carbonio.
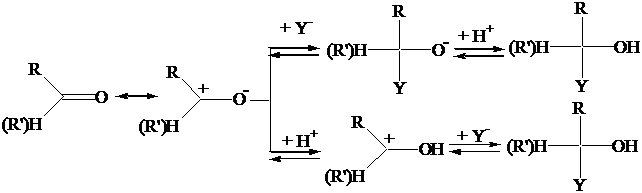
Le aldeidi sono più reattive dei chetoni per due effetti esercitati dal gruppo R: 1) effetto sterico, che sfavorisce l'attacco del nucleofilo Y-; 2) effetto induttivo +I, che diminuisce la carica positiva sul carbonio carbonilico.
Gli acidi carbossilici e i loro derivati subiscono reazione di sostituzione acilica. E' una reazione a due stadi: il 1° stadio è l'addizione nucleofila al carbonile; il 2° stadio è l'eliminazione dell'eteroatomo.
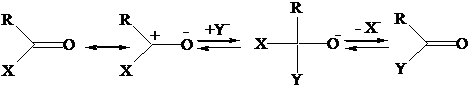
Come per aldeidi e chetoni la reazione è favorita da catalisi acida.

La reattività relativa delle varie classi di composti è governata principalmente dalla densità di carica positiva sull'atomo di carbonio carbonilico, influenzata dagli effetti -I e +M del sostituente X.
L'ordine di reattività è :

|
| Appunti su: 4 idrossifenolo, Sale di diazonio di un ammina, p-idrossifenolo ossidazione, grignard sostituzione nucleofile aromatiche, SH orto para orientanti, |
|
| Appunti Ingegneria tecnico |  |
| Tesine Geografia |  |
| Lezioni Biologia |  |